

The completion of the genome sequence of Drosophila melanogaster has provided a unique opportunity to initiate a new phase of systematic gene analysis of neuronal function. Although Drosophila as an experimental organism has made fundamental contributions to the fields of heredity and developmental biology, the field of neurobiology has also taken advantage of the powerful genetic techniques afforded by the fruitfly. Seymour Benzer and colleges identified and analyzed mutants with a variety of behavioral defects caused by single gene mutations. Such behavioral mutants initiated a wave of genetic studies into the function of the nervous system and led to the characterization of mutants such as Shibire and Shaker. These studies provided the foundation for a new generation of fly neurobiologists that employed systematic genetic screens for specific neurological phenotypes. The ease of performing electrophysiological analysis at Drosophila neuromuscular junctions readily complements the genetic approaches available in flies, making it an important model system for understanding synaptic function and development, as well as membrane excitability and neurological disease.
Using Drosophila as a model, the Littleton laboratory seeks to elucidate the molecular mechanisms underlying synapse formation, function and plasticity by combining molecular biology, protein biochemistry, electrophysiology and imaging approaches with genetics. In addition, studies are underway to characterize how the connections among neurons change to generate plasticity. The lab has identified many candidates for activity-dependent modulation of neuronal function and are determining how these genes contribute to cellular forms of behavioral plasticity. In addition, the lab is interested in the alterations in glial and neuronal signaling that contribute to epilepsy, Huntington’s disease and autism. Together, these approaches should expand our understanding of the basic mechanisms of synapse function and plasticity, as well as provide insights into plasticity changes that allow synaptic ensembles to alter the flow of information between synaptic partners.
Synaptic Transmission
Neurotransmitter release from synaptic vesicle fusion is the fundamental mechanism for neuronal communication at synapses. Since the work of Bernard Katz, it has been known that synaptic vesicle fusion is triggered by Ca2+ influx into the presynaptic terminal. Vesicular trafficking pathways are ubiquitous among eukaryotes, with fusion driven by the SNARE complex. Neurotransmitters can be released during evoked fusion following an action potential, or through spontaneous fusion of vesicles (termed “minis”) in the absence of nerve stimulation (Fig. 1). One of the major goals of my research program has been to define how vesicular trafficking mediates synaptic transmission, and how this process can be altered to change neuronal communication. At synapses, SNARE-mediated fusion is uniquely regulated to allow rapid and Ca2+-triggered synaptic vesicle fusion. Several key adaptations to the core fusion machinery require synapse specific SNARE-binding proteins, including Synaptotagmin 1 (Syt 1) and Complexin (Cpx). We have extensively studied how these proteins control synaptic output. Syt 1 is a synaptic vesicle protein that binds to SNARE complexes and membrane phospholipids in a Ca2+-dependent manner. My lab and others have show that Syt 1 functions as the Ca2+ sensor for fast synchronous neurotransmitter release. Cpx is a small cytosolic α-helical protein that binds assembled SNARE complexes. We have shown that association of Cpx with SNAREs allows it to function as a facilitator for synaptic vesicle fusion and as a fusion clamp to prevent premature exocytosis in the absence of Ca2+. A key avenue of our ongoing research program is to understand how these proteins, together with other key regulators like Munc18 and Munc13, regulate cycles of SNARE assembly and disassembly in a precisely coordinated fashion to mediate synaptic transmission. Similarly, we also seek to understand how the core fusion machinery can be rapidly modified, for example through phosphorylation, to alter presynaptic function and synaptic plasticity.
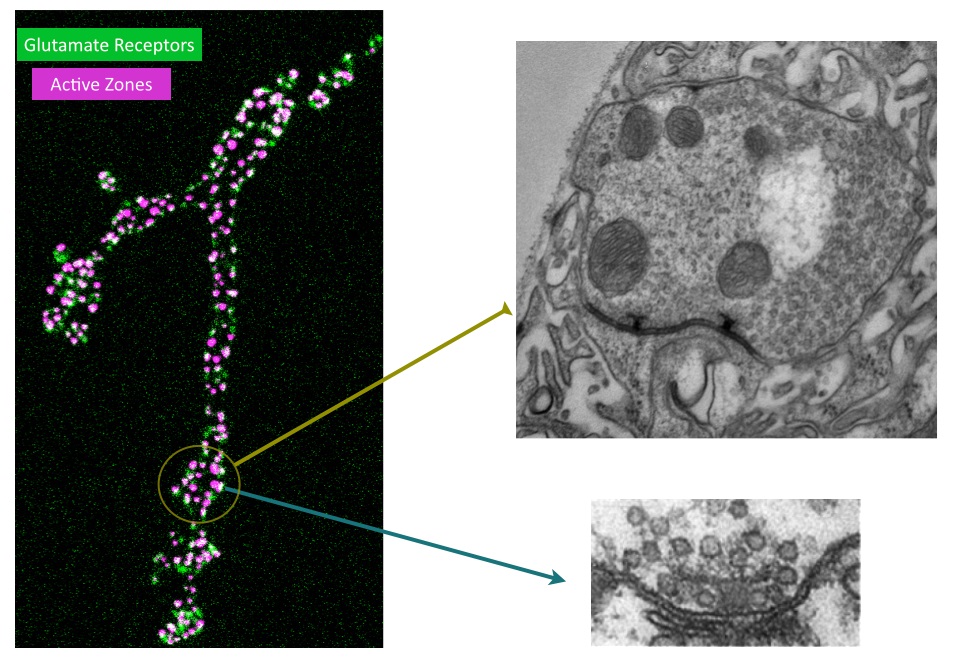
Although we have a research program in place to address how synapses form and function, I will highlight one approach below that provides a framework for how we use the Drosophila NMJ as a model synapse to uncover general principles of synapse biology. We have recently developed a new toolkit to optically visualize single synaptic vesicle fusion events from individual active zones. Evoked release following an action potential has been well characterized for its function in activating the postsynaptic cell, but the significance of spontaneous release is less clear. Whether receptors on the postsynaptic cell have the ability to distinguish between neurotransmitters released through the two independent fusion pathways is still under debate. If both vesicle release mechanisms activate the same set of receptors, crosstalk would occur between the two modes of fusion. However, if spontaneous release occurs at distinct release sites, the postsynaptic cell may be capable of differentiating between the two modes of release, suggesting spontaneous fusion may represent a separate information channel independent of the traditional Ca2+-activated evoked release pathway. The major confound to this question has been the inability to examine vesicle fusion at individual active zones. Classical electrophysiological studies of synaptic transmission measure the postsynaptic effect of neurotransmitter release over a population of release sites, precluding an analysis of how individual active zones participate in and regulate these two modes of vesicle fusion. We have developed transgenically expressed Ca2+ sensors that allow imaging of postsynaptic glutamate receptor activation following vesicle fusion, providing a mechanism to visualize all exocytotic events occurring through both spontaneous and evoked release pathways at Drosophila glutamatergic neuromuscular junctions. This toolkit allows us to generate release probability maps for both spontaneous and evoked fusion for all active zones at a synapse. By imaging a defined neuromuscular connection with ~ 300 active zones between the synaptic partners, we have begun to analyze the relationship between spontaneous and evoked release and determine how release mode and release probability are regulated at individual active zones at this model glutamatergic synapse. Unexpectedly, we have discovered that a subset of active zones are specifically dedicated to spontaneous release, indicating a population of postsynaptic receptors is uniquely activated by this mode of vesicle fusion. We have also found that release probability can vary more than 100-fold between neighboring active zones. This is an exciting finding and provides us a mechanism to understand how single active zone release probability is regulated. The ability to measure individual active zones for their release probability also allows us to determine how active zone release properties can change during synaptic plasticity. We hypothesize that evoked and spontaneous active zone release probability is not a static property of synaptic connections, and can be differentially regulated during distinct modes of synaptic plasticity. Synaptic plasticity mechanisms can be manifested through changes in N (the number of release sites), P (probability of release) or Q (quantal size). Many factors have been suggested to regulate these properties. However, the lack of a tool to examine single active zone release probability has made it difficult to dissect how these changes play out over a population of release sites. We are now in a position to examine how plastic changes in release properties at the NMJ map onto alterations in individual active zone release probability. By mapping single active zone release probability during both acute and homeostatic synaptic plasticity and following the properties, we can gain new insights into presynaptic mechanisms underlying synaptic communication and plasticity at a model glutamatergic synapse.The Drosophila NMJ contains spatially segregated active zones (termed T-bars) that oppose a distinct population of clustered glutamate receptors in the postsynaptic muscle (Fig. 2). This spatial arrangement of release sites and clustered receptors was used to analyze evoked release probability at individual release sites by measuring Ca2+ influx that occurs through postsynaptic glutamate receptors following vesicle fusion with a transgenic membrane-tethered (myristoylated) variant of the genetically encoded Ca2+-sensitive GFP (GCaMP6) that robustly detects postsynaptic responses to both evoked and spontaneous synaptic vesicle fusion events. We focused our initial analysis on NMJs at larval muscle fiber 4 which contains several hundred individual active zones (Fig. 2). Each synaptic bouton contains between 5 to 30 active zones that are spatially separated and opposed by a discrete cluster of glutamate receptors, providing a one-to-one match between the release machinery and the postsynaptic receptive unit. Transgenic lines expressing postsynaptic myrGCaMP show robust and spatially segregated signals for Ca2+ influx through glutamate receptors that occurs following synaptic vesicle fusion. Subsequent events at the same active zone produce similar Ca2+ profiles, allowing us to follow vesicle fusion at defined regions of interest (ROIs) over continuous imaging sessions for up to 30 minutes. Distinct ROIs defined by spatially segregated Ca2+ signals can be identified over the entire arbor, providing spatial information over time for release at all active zones. We observed that spontaneous Ca2+ signals detected optically are correlated with simultaneous electrophysiological recordings of minis at the NMJ for 99.8% of the events we detect. We find that spontaneous release probability per active zone is not uniform. Indeed, the distribution of spontaneous release probability is highly non-Poisson, with many low probability release sites, and a smaller subset of highly active release sites (Fig. 3).
In contrast to spontaneous fusion, evoked release is triggered following action potential propagation into presynaptic terminals and the influx of Ca2+ at active zones. The synchronous fusion of vesicles across many active zones is predicted to give rise to a postsynaptic current that underlies evoked synaptic transmission. Similar to spontaneous fusion, we were able to visualize evoked responses occurring at single ROIs, allowing a number of critical questions in synaptic function to be addressed. For example, do all active zones participate in both spontaneous and evoked fusion? Is release probability correlated between the two modes of vesicle fusion, and if not, is there a spatial logic to the organization of individual release sites along an axonal arbor? Do single synaptic vesicles fuse at individual active zones during an evoked response, or can multiple vesicles fuse from single active zones? Evoked signals were easily distinguished from spontaneous events during our imaging, as they were time locked to the stimulation. We observed that evoked release probability was not constant across all active zones, with most sites having a low probability. A small subset of highly active release sites was found in each preparation (Fig. 3). We also analyzed whether individual release sites participate in both evoked and spontaneous synaptic transmission by determining the number of evoked and spontaneous events that occurred per ROI during our imaging sessions. We observed that evoked and spontaneous fusion events could occur at the same site, indicating spontaneous and evoked vesicles can fuse from the same population of active zones. Although these dual mode active zones were the majority of sites we found, approximately 25% of the active zones showed only spontaneous release with no observed evoked responses. Given the presence of spontaneous only active zones, we examined whether evoked and spontaneous release probability would be correlated at mixed mode active zones, or if they would be independently regulated. We found that spontaneous and evoked release were highly non-correlated, indicating the presence of active zones with a high probability of evoked release and low probability of spontaneous release, and vise versa. These differences in release probability are easily noted in spatial heat maps displaying event rates per ROI for spontaneous and evoked responses at the same NMJ (Fig. 3), and indicate that spontaneous and evoked fusion are differentially regulated at individual active zones.
Moving forward, we will determine how evoked and spontaneous release probability is regulated at single active zones. We hypothesize that evoked active zone release probability will be primarily determined by presynaptic Ca2+ channel density and subsequent Ca2+ influx at single release sites. We will test this model and explore roles for synaptic vesicle properties and vesicle docking on release probability. For spontaneous only active zones, we hypothesize that these sites will either lack presynaptic Ca2+ channels, or have a greatly reduced density compared to active zones participating in evoked fusion. As observed in in the release probability maps in Fig. 3, individual active zones at the same NMJ connection have a wide distribution of evoked release probabilities, ranging form P=0.01 to P=0.35. There are several mechanisms that have been hypothesized to control release probability that we can directly examine. One hypothesis (Model 1) is that release probability is determined by the density of presynaptic N-type Ca2+ channels at a single active zone, thereby controlling the level of presynaptic Ca2+ entry. A higher density of Ca2+ channels would lead to increased Ca2+ levels at an active zone, and thereby increase the likelihood that the presynaptic release machinery would be activated. An alternative model (Model 2) is that presynaptic Ca2+ channel density and Ca2+ entry will be fairly constant over all active zones, and that evoked release probability is directly correlated with synaptic vesicle proteins and their number and/or state (i.e. phosphorylation status). It is well known that synaptic vesicles contain from 10-100 copies of many synaptic vesicle proteins, and that several of these can undergo post-translational modifications such as phosphorylation that may alter their function. By examining specific genetic manipulations alter single active zone release probability we can determine if specific synaptic proteins change release probability over all active zones while preserving a subset of active zones that show preferentially high release rates as observed in controls. Alternatively, if these proteins harbor determinants of release probability directly, we would expect to see that release probability would now decrease at all active zones and we would lose the preferred subset with high release rates altogether (as they would now lack this key determinant). Beyond these two models, it is possible that another determinant (Model 3) besides synaptic vesicle protein content/phosphorylation or active zone Ca2+ channel number/function regulates release probability. One such determinant could be the docking/priming state of synaptic vesicles at single release sites. Perhaps high P active zones have more docked synaptic vesicles, or larger pools of attached vesicles to the T-bar itself. As such, more vesicles of identical “protein content and state” would have access to a stochastic fusion process mediated by the same level of Ca2+ entry. Therefore, it would be very exciting to look for structural correlates for high P and low P active zones as well. The ability to map out single active zone release probability during various forms of synaptic plasticity paradigms will provide important new insights into fundamental mechanisms for how glutamatergic synapses transmit information.
Synapse Formation
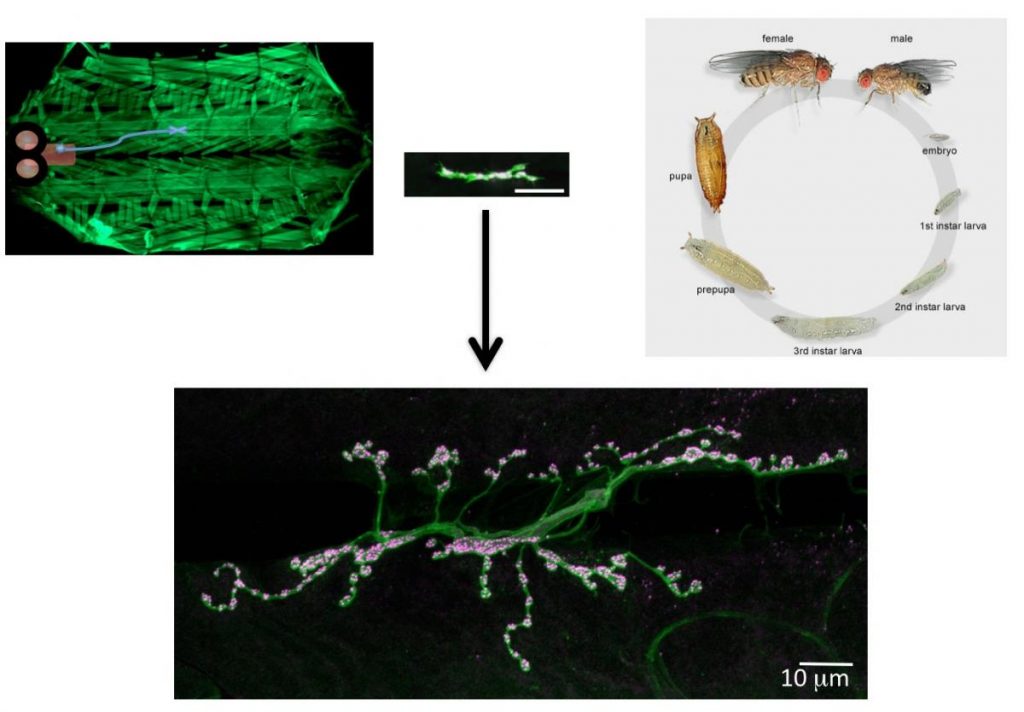
To identify neuronal pathways that control synapse formation and synaptic growth, we have conducted forward genetic screens for temperature-sensitive mutants that disrupt the process. Several Ca2+-dependent presynaptic proteins have been implicated in synaptic growth, but the source of Ca2+ and its regulation are unknown. In addition to retrograde signaling, we have found that presynaptic Ca2+ influx through N-type channels participates in synaptic growth via signaling pathways that are distinct from those that mediate neurotransmitter release. The opening of presynaptic N-type channels during robust synaptic activity allows presynaptic Ca2+ influx to modulate sprouting mechanisms that locally control synaptic remodeling. Linking presynaptic voltage-gated Ca2+ entry to downstream Ca2+-sensitive synaptic growth regulators provides an efficient activity-dependent mechanism for modifying synaptic strength.
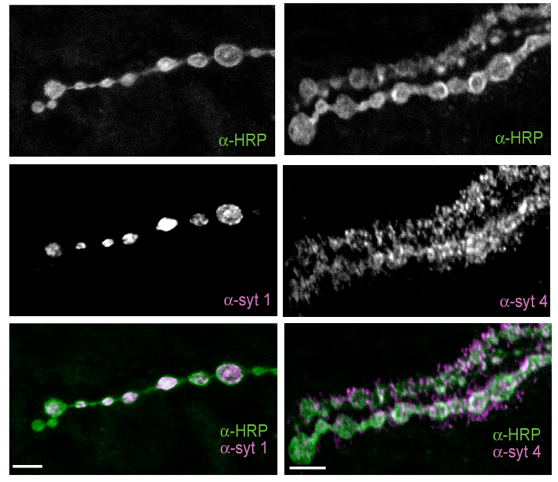
Several cell adhesion proteins and synaptic growth regulators reside adjacent to the active zone where presynaptic Ca2+ channels localize, in domains termed peri-active zones. We identified and characterized a peri-active zone signaling complex that controls synapse morphology and axonal branching through regulation of actin dynamics and endosomal trafficking of growth factors by the recycling endosome F-BAR/SH3 protein Nervous Wreck (NWK). The work on NWK was one of the first characterizations of a member of the now widely studied F-BAR protein family, a group of cytosolic proteins that bend membranes. NWK interacts with SNX16, which resides on early signaling endosomes and promotes synaptic growth through the Wingless and BMP pathways. These experiments defined a presynaptic trafficking pathway mediated by Snx16, NWK and the ESCRT complex that regulates synaptic growth signaling by modulating membrane dynamics and signal output at the interface between endosomal compartments.Recent findings from our lab and others have begun to identify the machinery that functions to sense Ca2+ rises in nerve terminals during activity and trigger information transfer between neurons through vesicle fusion and the release of neurotransmitters. Presynaptically, Ca2+ influx following stimulation drives a synchronous phase of SV fusion that occurs within milliseconds and a slower asynchronous component that can last for hundreds of milliseconds depending on the synapse and firing pattern of the neuron. Current models suggest Ca2+ influx drives SV fusion by binding to the conserved Synaptotagmin (Syt) family of vesicular Ca2+ sensors. Syt1 has been shown to function as the synchronous sensor, while its homolog Syt7 may act as the asynchronous sensor, although this model is still controversial. Postsynaptically, Ca2+ influx through neurotransmitter receptors or voltage-gated channels triggers synapse-specific plasticity. We recently discovered that Synaptotagmin 4 (Syt4) is an evolutionarily conserved Syt homolog that localizes to the postsynaptic compartment and regulates Ca2+-dependent postsynaptic vesicle trafficking and the release of retrograde signals that trigger synaptic growth and plasticity. In contrast to SV release, we know little about the regulation of postsynaptic vesicle fusion and how retrograde signals modulate synapse biology. By defining the molecular machinery for postsynaptic release, we hope to generate new models to advance the field of retrograde signaling into a more mechanistic analysis of its function. Using newly developed tools to visualize presynaptic and postsynaptic vesicle fusion, we can take advantage of genetic approaches in Drosophila to dissect synaptic signaling at a level of resolution few other model systems can offer.
Glial-Neuronal Signaling
We have recently begun to use Drosophila as a model organism to characterize how changes in glial Ca2+ oscillatory activity modify neuronal excitability, and how dysfunction of these pathways may contribute to epilepsy. Epilepsy is defined by incapacitating episodes of hypersynchronous neuronal firing, affecting 3 million Americans and over 50 million worldwide. Although a primary disruption of neuronal membrane excitability has been implicated in seizure pathogenesis in a subset of idiopathic epilepsy cases, the cellular events that trigger seizure initiation in most epilepsy patients are not well understood.
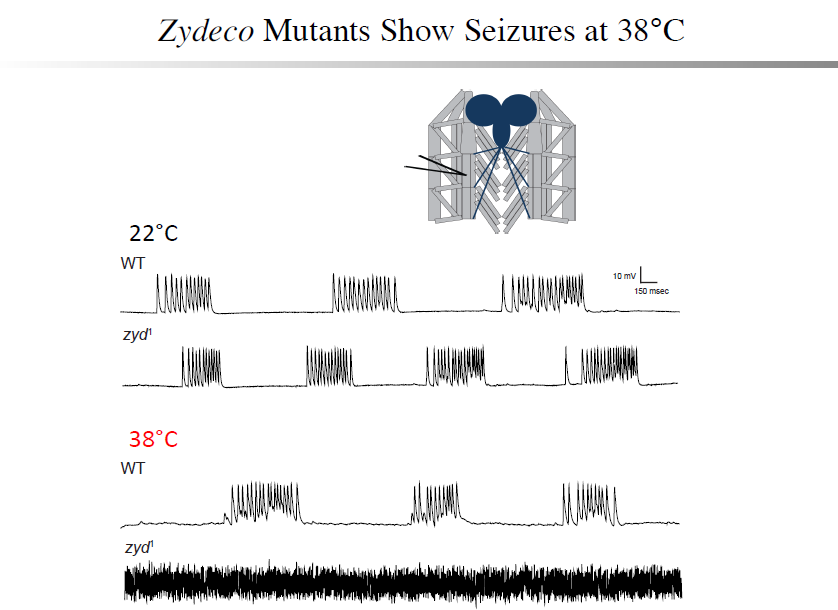
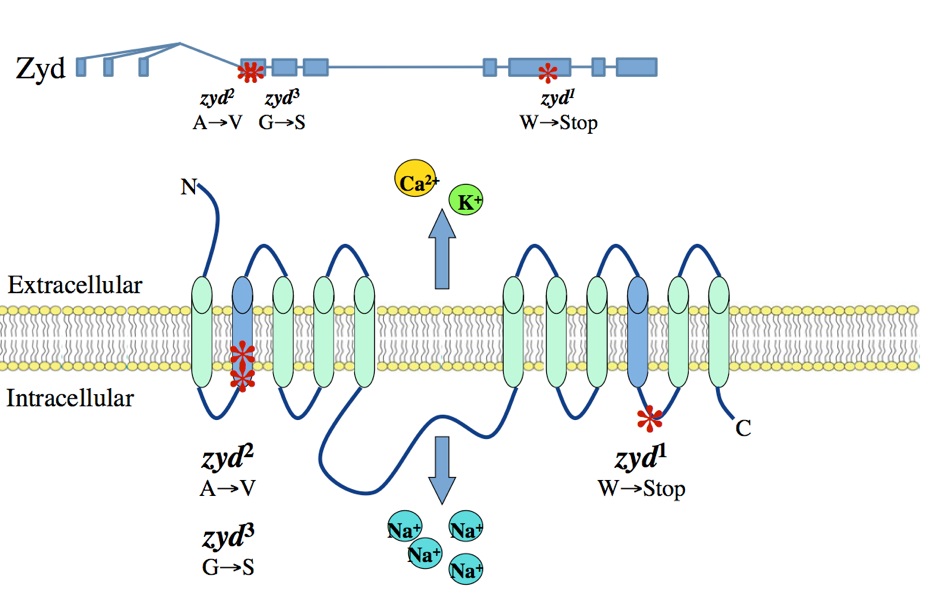
A non-neuronal origin for seizure induction has been suggested in some cases, with in situ studies correlating elevated glial Ca2+ oscillations with seizure initiation and in vivo work demonstrating several anti-epileptic drugs reduce glial Ca2+ oscillations. Although increased glial activity has been associated with abnormal neuronal excitability, the role of glia in the development and maintenance of seizures is poorly understood. Using an unbiased genetic screen for temperature-sensitive seizure mutants in Drosophila, we have identified and characterized a glial-specific gene that is required for microdomain glial Ca2+ oscillations. Acute disruption of this protein (termed Zydeco – an NCKX transporter), or acute induction of Ca2+ influx into glia via conditional TRP channels, induces the immediate onset of neuronal seizure activity. This is the first finding of conditional genetic manipulations of glial Ca2+ triggering neuronal seizures, indicating a Ca2+-dependent glial derived signal is important for maintaining appropriate neuronal excitability in the brain. Zydeco function is required specifically in cortex glia, which exhibit spatial segregation in Drosophila reminiscent of mammalian astrocytes, with each glial cell ensheathing multiple neuronal soma. Zydeco is not required in surface glial that constitute the blood-brain barrier or in ensheathing glia that insulate axons, and we have excluded a defect in ion balance as the cause of epileptic seizures in the mutant. These findings indicate that glial Ca2+ regulation is acutely involved in seizure generation, and that mutation of a glial-specific NCKX alters submembrane microdomain glial Ca2+ oscillations while increasing neuronal seizure susceptibility. We expect our studies will have significant impact on the current controversy concerning the physiological importance of glial Ca2+ signaling in the regulation of neuronal excitability. This debate on the significance of glial Ca2+ signaling is linked to the functional distinction between ER-mediated somatic Ca2+ oscillations and small, near-membrane Ca2+ oscillations in glial processes. Somatic glial Ca2+ waves are primarily mediated by the release of intracellular Ca2+ stores, and their relative importance in normal brain physiology has been disputed. Our findings indicate that microdomain glial Ca2+ activity is likely to be the more critical mode for glial modulation of neuronal function. We are currently characterizing the significance of both modes of glial Ca2+ oscillatory behavior to provide new insights into glial-neuronal communication and how non-neuronal triggers may contribute to epilepsy.
Epilepsy
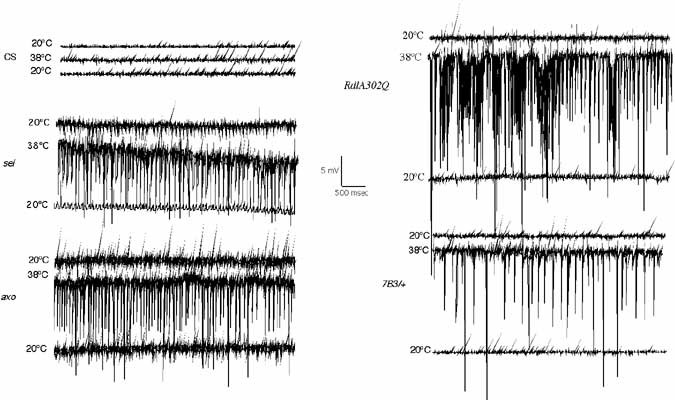
Epilepsy is a recurrent seizure syndrome caused by pathological synchronized firing of subpopulations of CNS neurons and is one of the most common neurological disorders, affecting approximately 3 million people within the US alone. Over 100 known Mendelian diseases include epilepsy as part of the neurological manifestation. Although epilepsy can be caused by many factors including fever, ischemic brain insults (stroke), traumatic brain injury or cancer, more than 40% of patients with epilepsy have been suggested to have a genetic component that contributes to the etiology. To identify neuronal dysfunctions that cause epilepsy, we have conducted behavioral screens for temperature-sensitive (TS) paralytic mutations induced by EMS that result in seizures. To date, 79 seizure-inducing mutations that define 27 complementation groups have been identified in large-scale screens of homozygous viable lines generated in the lab. We find that increasing neuronal activity drives overproliferation of synaptic connections, indicating activity-dependent rewiring occurs in Drosophila as observed in mammalian epilepsy models. To analyze transcriptional recoding during altered neuronal activity, we performed genome-wide DNA microarray analysis following multiple seizure induction and recovery paradigms in Drosophila mutants with acute or chronic alterations in neuronal activity. Approximately 250 genes implicated in cell adhesion, membrane excitability, and cellular signaling are differentially regulated, identifying a collection olfactivity-regulated transcripts that may link changes in neuronal firing patterns to transcription-dependent modulation of brain function. RNAi-mediated disruption or overexpression of several of these transcripts alters synaptic growth, suggesting an important role for transcriptional regulation in activity-dependent synaptic rewiring. These 250 activity-regulated transcripts represent exciting candidate genes that may regulate synaptic morphology and synaptic transmission, and will form the basis for new studies into how the brain responds to enhanced activity that occurs during plasticity, and in an exaggerated form during epilepsy.
Modeling Neurological Disease in Drosophila
Drosophila provides a power genetic system to model neurological disease and identify genetic and pharmacological interactions that reduce or eliminate pathology, with the expectation some of the pathogenic mechanisms that are uncovered may be conserved with those found in human patients. Many neurodegenerative diseases associated with protein misfolding have been modeled in Drosophila. These models replicate key neuropathological features characteristic of the diseases, such as late onset, progressive neurodegeneration, and formation of inclusions containing the mutant protein. Huntington’s Disease (HD) is an adult-onset dominant heritable disorder characterized by progressive psychiatric disruption, cognitive deficits, and loss of motor coordination. It is caused by expansion of a polyglutamine (polyQ) tract within the N-terminal domain of the Huntingtin (Htt) protein. The mutation confers a toxic gain-of-function phenotype, resulting in neurodegeneration. There is currently no cure, and the disease is fatal approximately 15 to 20 years after the age of onset. How polyQ expansion within Htt causes HD is poorly understood. We have generated Drosophila HD transgenic models expressing fluorescently tagged wildtype and pathogenic Htt proteins that allow for in vivo imaging of Htt localization, axonal transport and aggregate formation in live animals. We have identified several pathologies associated with Htt polyQ expression in fly neurons, and have attempted to identify small molecules and genetic interactors that can revert HD pathology in our system. We are also studying mutants of the Drosophila Htt homolog to define the normal function of Htt within neurons.
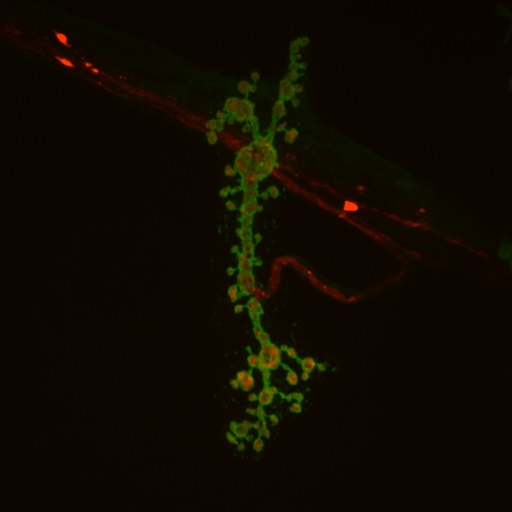
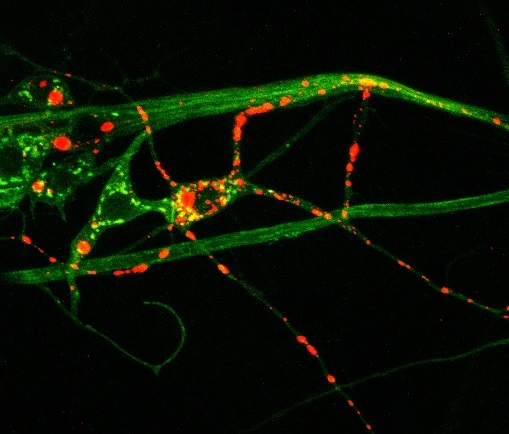
Above: Huntingtin protein (red) aggregates inside neuronal processes (green)
Autism Spectrum Disorder
In contrast to the neurodegenerative diseases, whether Drosophila can provide an effective model system for neurodevelopmental diseases like autism spectrum disorders (ASDs) is still an open question. Defining molecular pathways that are dysfunctional in ASDs is key to understanding their pathogenesis. In Alzheimer’s and Parkinson’s Disease, identification of single gene mutations in the 5-10% of genetic cases have revealed core molecular pathways that are altered, including in the larger category of sporadic cases. A major question is whether a similar molecular pathway will emerge for autism based on the recent identification of defined mutations and de novo genome copy number variations that account for 10-20% of ASDs. Current evidence suggests the disease may result from disruption in synapse formation and synaptic plasticity during development. We are taking advantage of genetic manipulations available in Drosophila to explore the mechanisms by which newly identified autism-associated synaptic proteins may alter neuronal activity or synaptic connectivity. We will use these models to explore how basic synaptic properties may be disrupted in autism-associated genes implicated from human genetic studies.